10
Mar
If you’re searching for star rna seq aligner images information connected with to the star rna seq aligner keyword, you have come to the right site. Our website frequently provides you with hints for seeing the maximum quality video and image content, please kindly search and locate more enlightening video content and images that match your interests.
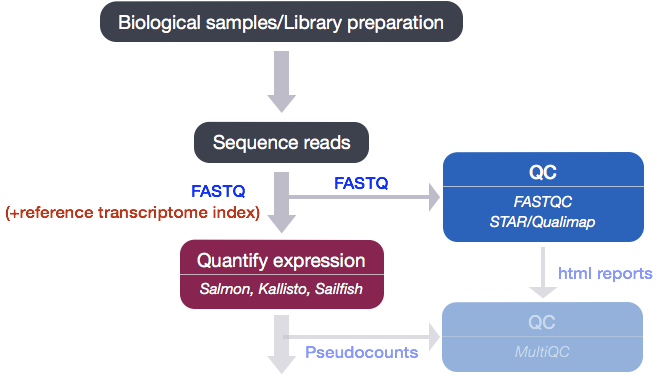
Star Rna Seq Aligner. STAR maps the reads to the genome and writes several output les such as alignments SAMBAM mapping summary statistics. Different parts of a read can be mapped to different. Aligning RNA-seq data The theory behind aligning RNA sequence data is essentially the same as discussed earlier in the book with one caveat. STAR –option1-name option1-values–option2-name option2-values.

Then Cuffmerge merges the previous assembles of individual condition. STAR outputs read counts per gene into PREFIXReadsPerGeneouttab file with 4 columns which correspond to different strandedness options. Read alignment quality control and data analysis were performed using Visualization Pipeline for RNA-seq VIPER 67. To accurately estimate the abundance of each transcript any possible isoforms are considered in this step. BioCloud RNA-Seq STAR Result Documentation. 79 Arriba can also detect viral integration sites internal tandem duplications whole exon duplications circular RNAs enhancer hijacking events involving immunoglobulinT-cell receptor loci and.
Gene ID column 2. STAR –option1-name option1-values–option2-name option2-values. STAR is a fast RNA-Seq read mapper with support for splice-junction and fusion read detection and it was designed to align non-contiguous sequences directly to a reference genome. Generate a genome index using genome reference information. Gene models in Eukaryotes contain introns which are often spliced out during transcription. STAR can align spliced sequences of any length with moderate error rates providing scalability for.
Previous post
Starting parameters trochoidal milling forumNext post
Stages of peach growth